This code implements methods for mapping a disease gene in a case and control design study, using only multilocus estimated allele frequency data as might be obtained from DNA pools. Because there is essentially no phase or linkage information in multilocus allele frequency data, and because of the noise in allele frequency estimates obtained from DNA pools, this approach will give less information than an analysis of haplotype or genotype data. However, typing DNA pools is very economical and the laboratory effort saved might allow a larger number of markers to be typed. It remains unclear what the optimal experimental strategy is.
Full details of the statistical methods implemented are given in the following papers.
For instructions on using the program, consult this manual page ( HTML | PDF )
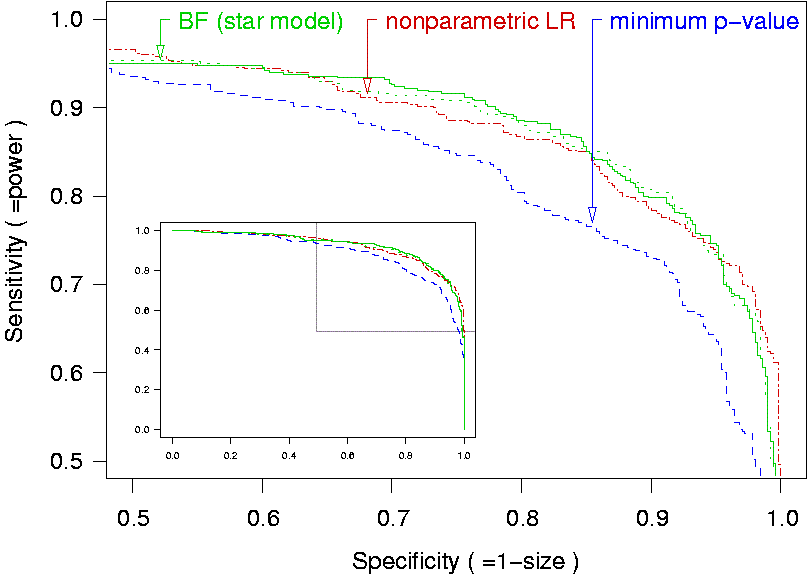
The code can compute two test statistics, a Bayes factor (green curve) and a nonparametric likelihood ratio (red curve), for detecting the presence of a causal variant or QTL. Both tests are more powerful than classical single point methods (blue curve), as shown in the ROC curves below.
The nonparametric likelihood ratio can be computed very quickly, requiring time that is linear in number of SNPs and linear in number of cases. Both tests can be performed without using MCMC methods, so there is no need to worry about burn-in or mixing. These approaches are therefore suitable for use in permutation tests or simulation approaches for assessing significance.
Several methods for inferring the position of a causal variant or QTL are implemented. These produce point estimates of position that are better than picking the map position of the marker with the smallest p-value. Well calibrated credibility intervals for position can also be computed.
The original POOLMAP program that was used to generate results for the Annals of Human Genetics paper is available on request, but all its functionality and much more is now provided by the poolmapB program.
I would like to keep track of who has downloaded the program so that I can inform you about any bugs that I subsequently find, and for various grant-related purposes. To this end, please enter your email address, and then click on the 'Download' button.